Argo Workflows is an open source, Kubernetes-native workflow engine for automating multi-step gene computing pipelines. This guide shows you how to run a complete Burrows-Wheeler Aligner (BWA) sequence alignment pipeline on an Alibaba Cloud ACK One distributed workflow cluster.
Background
Gene computing workflows
A gene computing workflow is a sequence of computational tasks designed to achieve a specific analysis goal in genomics research. Typical workflows include data preprocessing, sequence alignment, variant calling, gene expression analysis, and phylogenetic tree construction—each step producing output that feeds the next.
Why Argo Workflows for gene computing
Argo Workflows handles the complexity of multi-step genomic pipelines in two ways:
-
Containerized execution: Each analysis step runs in its own Docker container, ensuring consistent results across environments and eliminating tool dependency conflicts.
-
Flexible orchestration: Workflows can branch conditionally, run steps in parallel, and loop over large datasets—all defined in a single YAML file.
Challenges with open-source Argo Workflows
Running Argo Workflows at scale in genomics introduces three operational challenges:
-
Cluster operations: Optimizing and maintaining a large Kubernetes cluster requires deep infrastructure expertise that most research teams lack.
-
Scale: Scientific experiments often involve vast parameter spaces and thousands of concurrent jobs, pushing beyond what standard open-source configurations can handle.
-
Resource efficiency: Gene data analysis is resource-intensive. Intelligently scheduling workloads and scaling compute on demand requires additional tooling on top of open-source Argo.
The Alibaba Cloud ACK One team built the distributed workflow Argo-based cluster to address these challenges directly.
Distributed workflow Argo-based cluster
The distributed workflow Argo-based cluster is built on open-source Argo Workflows with a serverless model: workflows run on Elastic Container Instance (ECI) rather than long-lived nodes. This design provides:
-
Elastic scaling: Kubernetes cluster parameters are optimized for large-scale job scheduling. Compute capacity scales up and down automatically with your workload.
-
Cost efficiency: Workflows use preemptible ECI instances to reduce compute costs.
-
Rich execution strategies: Concurrency, loops, retries, and DAG dependencies are all supported—covering the full range of gene computing orchestration needs.

Argo Workflows is widely used in gene computing, autonomous driving, and financial simulation. To discuss best practices with the ACK One team, join their DingTalk group (ID: 35688562).
Run a BWA sequence alignment workflow
This example implements a classic BWA sequence alignment pipeline with three stages. BWA sequence alignment maps raw sequencing reads back to a reference genome—the first step in identifying genetic variants.
| Stage | What it does |
|---|---|
| bwaprepare | Downloads and decompresses the FASTQ sequencing files and reference genome, then builds the BWA index |
| bwamap | Aligns the sequencing reads to the reference genome; runs in parallel for each FASTQ file |
| bwaindex | Generates paired-end alignments, then creates, sorts, and indexes the final BAM file |
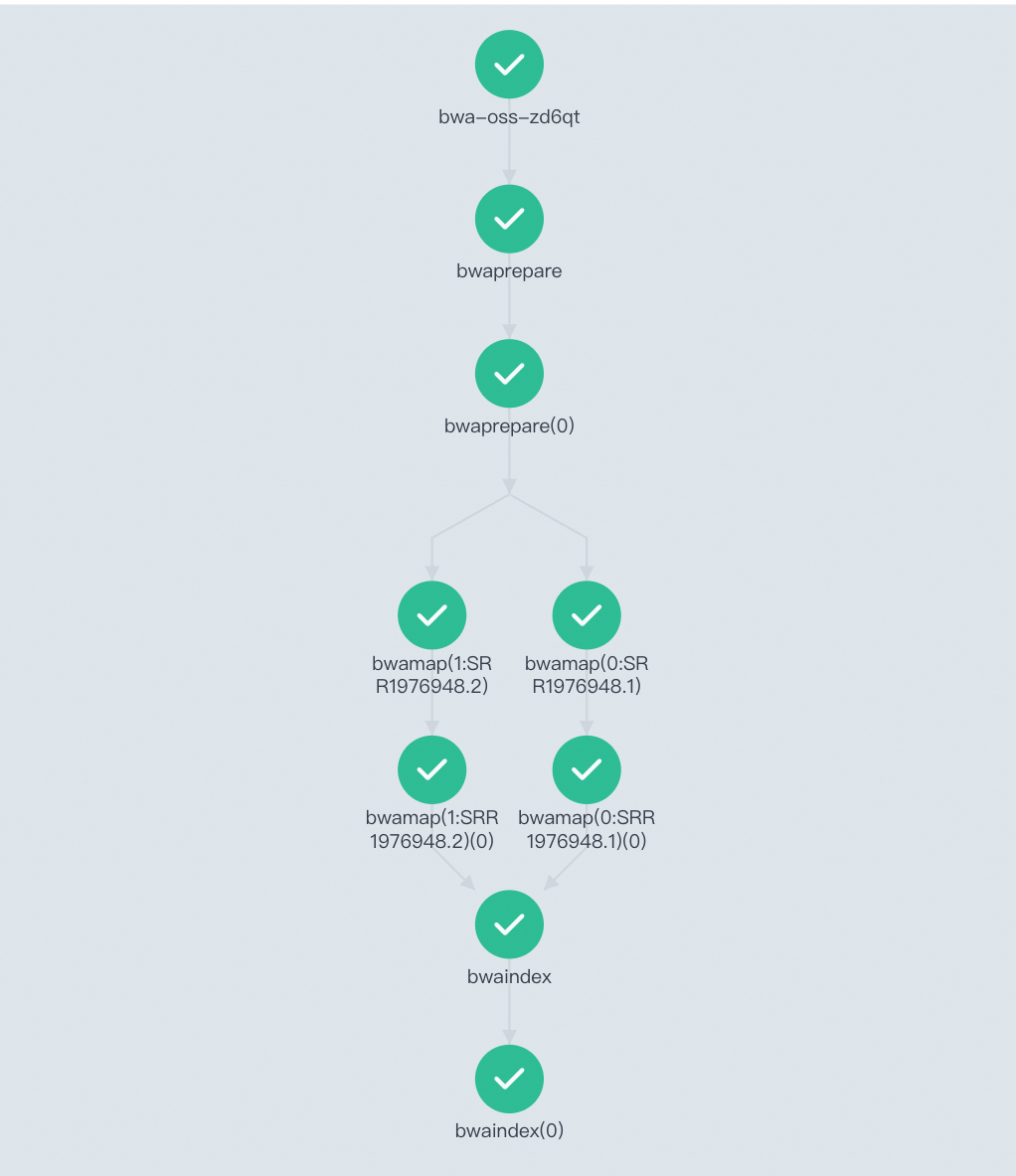
The three stages run as a Directed Acyclic Graph (DAG): bwaprepare must complete before bwamap starts, and bwamap must complete before bwaindex starts. Within bwamap, the two FASTQ files are processed in parallel.
Prerequisites
Before you begin, ensure that you have:
-
A distributed workflow Argo-based cluster. See Create a distributed workflow Argo-based cluster.
-
Argo Server access enabled for the cluster. See Enable Argo Server to access the workflow cluster.
-
An Object Storage Service (OSS) volume mounted so the workflow can access OSS files as if they were local files. See Use a volume. The persistent volume claim (PVC) name for this example is
pvc-oss.
Create and run the workflow
Create the workflow using the following YAML. For details on submitting a workflow, see Create a workflow.
To use this workflow with your own data, update the four parameters at the top of the arguments block:
| Parameter | Description | Example value |
|---|---|---|
fastqFolder |
Directory on the mounted OSS volume where files are stored | /gene |
reference |
URL of your reference genome (.fa.gz) |
https://ags-public.oss-cn-beijing.aliyuncs.com/alignment/subset_assembly.fa.gz |
fastq1 |
URL of the first FASTQ sequencing file | https://ags-public.oss-cn-beijing.aliyuncs.com/alignment/SRR1976948_1.fastq.gz |
fastq2 |
URL of the second FASTQ sequencing file | https://ags-public.oss-cn-beijing.aliyuncs.com/alignment/SRR1976948_2.fastq.gz |
The rest of the YAML defines the pipeline logic and stays the same for any dataset.
apiVersion: argoproj.io/v1alpha1
kind: Workflow
metadata:
generateName: bwa-oss-
spec:
entrypoint: bwa-oss
arguments:
parameters:
- name: fastqFolder # Directory on the OSS volume where files are saved.
value: /gene
- name: reference # Reference genome file URL.
value: https://ags-public.oss-cn-beijing.aliyuncs.com/alignment/subset_assembly.fa.gz
- name: fastq1 # First raw sequencing data file URL.
value: https://ags-public.oss-cn-beijing.aliyuncs.com/alignment/SRR1976948_1.fastq.gz
- name: fastq2 # Second raw sequencing data file URL.
value: https://ags-public.oss-cn-beijing.aliyuncs.com/alignment/SRR1976948_2.fastq.gz
volumes: # Mount the OSS volume so all stages share the same storage.
- name: ossdir
persistentVolumeClaim:
claimName: pvc-oss
templates:
- name: bwaprepare # Stage 1: Download files and build the reference genome index.
container:
image: registry.cn-beijing.aliyuncs.com/geno/alltools:v0.2
imagePullPolicy: Always
command: [sh,-c]
args:
- mkdir -p /bwa{{workflow.parameters.fastqFolder}}; cd /bwa{{workflow.parameters.fastqFolder}}; rm -rf SRR1976948*;
wget {{workflow.parameters.reference}};
wget {{workflow.parameters.fastq1}};
wget {{workflow.parameters.fastq2}};
gzip -d subset_assembly.fa.gz;
gunzip -c SRR1976948_1.fastq.gz | head -800000 > SRR1976948.1;
gunzip -c SRR1976948_2.fastq.gz | head -800000 > SRR1976948.2;
bwa index subset_assembly.fa;
volumeMounts:
- name: ossdir
mountPath: /bwa
retryStrategy: # Retry up to 3 times to handle transient network failures during file downloads.
limit: 3
- name: bwamap # Stage 2: Align one FASTQ file to the reference genome.
inputs:
parameters:
- name: object
container:
image: registry.cn-beijing.aliyuncs.com/geno/alltools:v0.2
imagePullPolicy: Always
command:
- sh
- -c
args:
- cd /bwa{{workflow.parameters.fastqFolder}};
bwa aln subset_assembly.fa {{inputs.parameters.object}} > {{inputs.parameters.object}}.untrimmed.sai;
volumeMounts:
- name: ossdir
mountPath: /bwa
retryStrategy:
limit: 3
- name: bwaindex # Stage 3: Generate paired-end alignments and produce the final sorted BAM file.
container:
command:
- sh
- -c
args:
- cd /bwa{{workflow.parameters.fastqFolder}};
bwa sampe subset_assembly.fa SRR1976948.1.untrimmed.sai SRR1976948.2.untrimmed.sai SRR1976948.1 SRR1976948.2 > SRR1976948.untrimmed.sam;
samtools import subset_assembly.fa SRR1976948.untrimmed.sam SRR1976948.untrimmed.sam.bam;
samtools sort SRR1976948.untrimmed.sam.bam -o SRR1976948.untrimmed.sam.bam.sorted.bam;
samtools index SRR1976948.untrimmed.sam.bam.sorted.bam;
samtools tview SRR1976948.untrimmed.sam.bam.sorted.bam subset_assembly.fa -p k99_13588:1000 -d T;
image: registry.cn-beijing.aliyuncs.com/geno/alltools:v0.2
imagePullPolicy: Always
volumeMounts:
- mountPath: /bwa/
name: ossdir
retryStrategy:
limit: 3
- name: bwa-oss # DAG orchestrator: defines stage dependencies and parallel execution.
dag:
tasks:
- name: bwaprepare
template: bwaprepare
- name: bwamap
template: bwamap
dependencies: [bwaprepare] # Runs after bwaprepare completes.
arguments:
parameters:
- name: object
value: "{{item}}"
withItems: ["SRR1976948.1","SRR1976948.2"] # Processes both files in parallel.
- name: bwaindex
template: bwaindex
dependencies: [bwamap] # Runs after both bwamap tasks complete.Verify the results
After the workflow completes, check the results in two places:
-
Open the Workflow Console (Argo) to view the DAG and confirm all stages succeeded.
-
Log in to the OSS console and find the alignment result files in your OSS folder. The final output is
SRR1976948.untrimmed.sam.bam.sorted.bamand its index.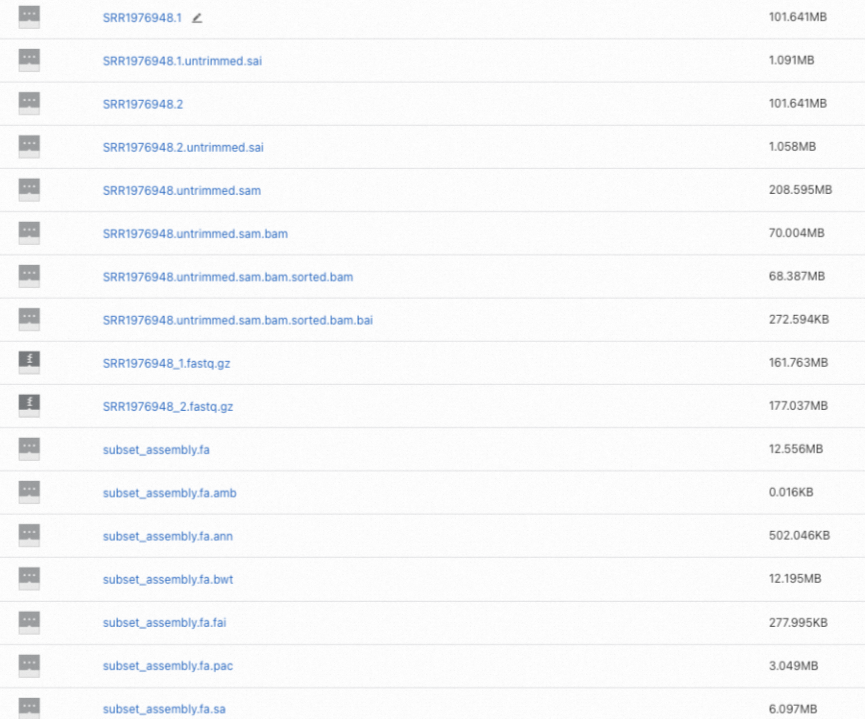
What's next
-
For an overview of distributed workflow Argo-based clusters, see Overview of distributed workflow Argo-based clusters.
-
To learn more about Argo Workflows features such as loops, DAGs, and retry strategies, see the Argo Workflows documentation.