AnalyticDB for MySQLは、クラウドでホストされるデータウェアハウジングサービスで、ペタバイト単位のデータを高い同時実行性と低いレイテンシで処理できます。 遺伝子検索システムは、AnalyticDB for MySQLのベクター検索機能に基づいて構築されています。 このシステムは、数十億のベクトルデータ記録に対して数ミリ秒以内にクエリおよび分析を実行することができ、肺炎ウイルスの予防および制御、ならびに治療薬および関連ワクチンの研究開発の効率を向上させる。
遺伝子配列検索技術のシナリオと現状
遺伝子配列検索技術は、以下のシナリオで使用されます。
肺炎ウイルスの接触追跡と分析により、ウイルス宿主の特定と効果的な予防および管理対策の実施を支援します。
ウイルスの複製と感染の分析、治療薬とワクチンの開発を支援します。
肺炎ウイルスに類似したウイルスの遺伝子配列の検索。
ウイルスが急速に拡散するにつれて、効率的なマッチングアルゴリズムが、遺伝子配列検索のために緊急に必要とされる。 これに関連して、AnalyticDB for MySQL技術チームは遺伝子断片を1024次元の特徴ベクトルに変換しました。これは、2つの遺伝子断片を照合するプロセスが2つのベクトル間の距離の計算に変換されることを意味します。 これにより、コンピューティングオーバーヘッドを削減し、結果を返すのに必要な時間をミリ秒に短縮できます。 このプロセスは、遺伝子断片の予備スクリーニングに使用することができる。 次に、遺伝子類似性計算のBLASTアルゴリズムを使用して、正確な類似性ランキングを生成し、より効率的な方法で遺伝子配列のマッチング計算を完了する。 マッチングアルゴリズムの複雑さは、O(M + N) からO(1) に低減される。 AnalyticDB for MySQLは、強力な機械学習分析ツールも提供します。 これらのツールは、遺伝子からベクターへの技術を介して、局所および疾患関連標的遺伝子断片を特徴ベクターに変換することができる。 次に、これらのベクターを遺伝子薬の研究開発に使用して、遺伝子分析のプロセスを加速することができます。
AnalyticDB for MySQLの遺伝子検索システム
肺炎ウイルスのRNA配列は、塩基配列とも呼ばれる一連のヌクレオチド配列として表すことができる。 RNA配列は、アデニン、シトシン、グアニン、およびチミンについてA、C、G、およびTと標識された4つのヌクレオチドから構成される。 各文字は塩基を表し、これらの塩基は隙間なく互いに連結されている。 それぞれの種には独自のRNA配列がありますが、パターンを見つけることができます。 遺伝子検索システムは、システムに提出されたものと類似の遺伝子を検索し、特定のウイルスのRNA配列を分析することができる。
AnalyticDB for MySQLを使用して遺伝子フラグメントを取得する方法を示すために、AnalyticDB for MySQL技術チームは、GenBankから大量のウイルスRNAフラグメントデータと、GenBankおよびGoogle Scholarからのウイルス関連論文をAnalyticDB for MySQL遺伝子検索データベースにインポートしました。
次に、技術チームは肺炎ウイルス配列をAnalyticDB for MySQLの遺伝子検索システムにアップロードしました。 ミリ秒後、0.8を超える一致度を有する類似の遺伝子断片が返還され、これには、パゴリンによって運ばれる肺炎ウイルス (GD/P1L) 、コウモリによって運ばれる肺炎ウイルス (RaTG13) 、SARSウイルス、およびMARSウイルスが含まれる。 GD/P1Lは、0.974のマッチ度を有する最良の配列マッチであった。 肺炎ウイルスはパンゴリンを介して人々に伝染したと推測されています。
RNA断片が非常に類似している場合、2つのRNA配列は、類似のタンパク質発現および構造を有し得る。 肺炎ウイルスとSARSウイルスの一致度、および肺炎ウイルスとMARSウイルスの一致度は0.8を超えています。 これは、SARSまたはMARSウイルスのいくつかの研究結果を使用して、肺炎ウイルスをよりよく理解できることを示しています。 システムは、一致した各ウイルスに関する学術論文を取得し、これらの論文をテキスト分類アルゴリズムによって試験、ワクチン、および投薬のカテゴリに分割する。です。One of the testing methods for SARS is fluorescence quantitative PCR detection. この方法は、肺炎ウイルスをテストするために使用されます。 遺伝子ワクチンおよび免疫応答のin vivo誘導法は開発中である。 レムデシビルおよび関連インターフェロンは、肺炎ウイルス患者を治療するために使用される。
アーキテクチャ
AnalyticDB for MySQL is used in the gene retrieval system to store and query feature vectors produced for gene sequences and all structured data such as gene sequence lengths that contain academic paper names, gene types, and DNA or RNA. During the query process, a gene vector extraction model is used to convert genes into vectors and perform coarse sorting retrieval in the AnalyticDB for MySQL vector database. In the vector matching result set, the BLAST algorithm is used to perform precise sorting and return the most similar gene sequences.
The core of the gene retrieval system of AnalyticDB for MySQL is the gene vector extraction model. This model can convert nucleotide sequences into vectors. AnalyticDB for MySQL extracts and trains all the RNA sequence samples of a variety of viruses to help the model better calculate the RNA similarity between viruses. The gene vector extraction model can be easily extended to genes of other species.
遺伝子ベクトル抽出アルゴリズム
単語ベクトル技術は、機械翻訳、読解、意味解析などの分野ですでに広く実装されており、大成功を収めています。 単語ベクトル化は、単語の意味を表現するために分布意味論的アプローチを使用する。 単語の意味はその文脈にあります。 単語バンク内の単語を使用して、段落内の不足している単語を入力する必要があるテストに戻ります。 これらのテストでは、単語のコンテキストは、単語自体を正確に反映することができる。 正しい単語を選択すると、空いている単語の意味を理解していることを示します。 したがって、単語ベクトルアルゴリズムは、所与の単語と周囲の単語との関係を介して、テキスト内の各単語のベクトルを生成することができる。 次に、単語ベクトルの類似度を計算して単語間の類似度を求めることができる。
同様に、遺伝子配列は特定の規則に従い、遺伝子配列の各部分は異なる機能性および意味を表す。 長い遺伝子配列は、研究目的でより小さな単位 (「単語」) に分割することができます。 これらの「単語」には、相互接続され、相互作用して対応する機能を完成させ、表現を形成するため、コンテキストもあります。 生物科学者は、遺伝子配列単位をベクトル化するためにワードベクターアルゴリズムを使用します。 2つの遺伝子ユニット間の高い類似性は、両方の遺伝子ユニットが常に一緒に現れ、一緒に機能を発現することを示しています。
通常、AnalyticDB for MySQLの遺伝子ベクター抽出アルゴリズムには、次の手順が含まれます。
アミノ酸配列で単語を定義します。
バイオインフォマティクス分野では、アミノ酸配列を分析するためにkマーが使用される。 K量体は、ヌクレオチド配列がK塩基を含むストリングに分割された後に得られる。 これは、連続ヌクレオチド配列から長さK塩基の配列を繰り返し選択することによって行われる。 ヌクレオチド配列の長さがLである場合、以下の数のk − merを得ることができる: L − K + 1。 配列の長さが12であり、k − merの長さが8である場合、5つの8 − merは、以下の式: 12 − 8 + 1から得ることができる。 これらのkマーは、アミノ酸配列中の「ワード」と等価である。
アミノ酸配列のコンテキストを見つけ、遺伝子配列の「単語」を1024次元ベクターに変換します。
コンテキストは、単語ベクトルアルゴリズムにおいて重要な役割を果たす。 AnalyticDB for MySQLの遺伝子ベクター抽出アルゴリズムは、アミノ酸断片から長さLのウィンドウを選択します。 このウィンドウ内のアミノ酸断片は、同じ文脈内にあると考えられる。 例えば、ヌクレオチド配列CTGGATGAについて長さ10のウィンドウが選択される場合、AnalyticDB For MySQLの遺伝子ベクター抽出アルゴリズムは、CTGGATGAを以下の5マーに変換する: AACTG、ACTGG、CTGGA、GGATG、およびGATGA。 CTGGAの場合、他の4つの5-merがそのコンテキストを構成します。 AnalyticDB for MySQLの遺伝子ベクトル抽出アルゴリズムは、単語ベクトル空間トレーニングモデルを使用して、既存の遺伝的kマーをトレーニングし、kマーを1024次元ベクトルに変換します。
単語ベクトルモデルと同様に、k − merベクトルモデルもベクトルに対して数学的計算を実行する。
ベクトル減算:
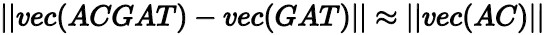
ベクトル付加:
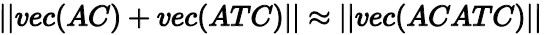
ベクトル減算式は、「ACGATベクトル − GATベクトル」とACベクトルとの間の距離が非常に近いことを示す。 ベクトル加算式は、「ACベクトル + ATCベクトル」とACATCベクトルとの間の距離も非常に近いことを示す。 長いアミノ酸配列のベクターを計算する場合、これらの数学的特徴に基づいて、各断片のk − mer配列をこの配列に付加することができる。 次に、結果を正規化して、アミノ酸配列全体のベクターを得ることができます。 このアプローチの精度を向上させるには、遺伝子フラグメントをテキストフラグメントと見なし、doc2vecを使用してシーケンス全体を計算用のベクトルに変換します。 アルゴリズムの性能を検証するために、AnalyticDB for MySQLの遺伝子ベクター抽出アルゴリズムは、BLASTアルゴリズム配列とベクター − 遺伝子間距離配列との間の類似性を計算する。 両シーケンスのスピアマン順位相関係数は0.839である。 これは、DNA配列をベクターに変換することが、類似の遺伝子断片の予備スクリーニングの有効な方法であることを示す。
ベクトル検索の概要
ベクトル検索を含む一般的なアプリケーションシステムでは、開発者はFaissなどのベクトル検索エンジンを使用してベクトルデータを格納し、次にリレーショナルデータベースを使用して構造化データを格納します。 2つのシステムを使用して異なるデータを照会する必要があります。 この解決策は余分な開発努力を必要とし、最適なデータクエリ性能を提供しない。
AnalyticDB for MySQLは、クラウドでホストされるデータウェアハウジングサービスで、ペタバイト単位のデータを高い同時実行性と低いレイテンシで処理できます。 数十億のベクトルデータレコードに対してミリ秒以内にクエリを実行し、100ミリ秒以内に応答を返すことができます。 AnalyticDB for MySQLは、MySQLプロトコルおよびSQL:2003構文と完全に互換性があります。 これは、画像、テキスト推奨、声紋、およびヌクレオチド配列の類似性照会および分析をサポートするベクトル検索機能を提供する。 AnalyticDB for MySQLは、複数の都市のセキュリティプロジェクトで広く使用されています。
AnalyticDB for MySQLは、構造化データと非構造化データの取得と分析をサポートしています。 SQLインターフェイスを使用して、遺伝子検索システムや遺伝子および構造化データのハイブリッド検索システムなどのシステムを構築できます。 ハイブリッド検索シナリオでは、AnalyticDB for MySQLのオプティマイザは、データ分布とクエリ条件に基づいて最適な実行プランを選択し、リコール率を確保しながら最適なパフォーマンスを実現します。 たとえば、次のSQL文を使用してRNAヌクレオチド配列を取得できます。
-- Query gene segments that are similar to the submitted sequence vectors within the RNA sequence.
select title, # The article name.
length, # (#) The gene length.
type, # mRNA or DNA.
l2_distance(feature, array[-0.017,-0.032,...]::real[]) as distance # The vector distance.
from demo.paper a, demo.dna_feature b
where a.id = b.id
order by distance; # Sort by vector similarity.上記のSQL文では、demo.paperテーブルにはアップロードされた各記事の基本情報が格納され、demo.dna_featureテーブルには各種の遺伝子配列に対応するベクターが格納されます。 遺伝子対ベクターモデルは、遺伝子を [-0.017、-0.032、...] などのベクターに変換するために使用され、これらのベクターは、AnalyticDB for MySQLデータベースでの検索に使用できます。
本システムはまた、構造化および非構造化情報 (ヌクレオチド配列) のハイブリッド検索をサポートする。 たとえば、肺炎ウイルスに似ている遺伝子セグメントをクエリするには、SQL文にwhere title like '% COVID-19 %' を追加するだけです。
付録
[1] Mikolov Tomas; et al。 (2013) 。 「ベクトル空間における単語表現の効率的な推定」。 arXiv:1301.3781。
[2] Mikolov Tomas、Sutskever Ilya、Chen Kai、Corrado、Greg S. 、Dean Jeff (2013) 。 単語やフレーズの分散表現とその構成。 神経情報処理システムの進歩。 arXiv:1310.4546。 ビブコード: 2013arXiv1310.4546M。
[3] メープルソンダニエル、ガルシアアクシネッリ、ゴンザロ、ケトルボロージョージ、ライトジョナサンとクラビホ、ベルナルドJ. (2016) 。 「KAT: NGSデータセットとゲノムアセンブリを品質管理するためのKマー分析ツールキット」。 バイオインフォマティクス。 33(4): 574-576。 doi:10.1093 /バイオインフォマティクス /btw663。 ISSN 1367-4803。 PMC 5408915。 PMID 27797770。
[4] Quoc LeとTomas Mikolov。 (2014) 。 文と文書の分散表現。 機械学習に関する国際会議では、1188〜1196ページ。
[5] ヒトゲノムHG38、https://hgdownload.cse.ucsc.edu/goldenPath/hg38/bigZips/hg38.chromFa.tar.gz 。
[7] ジュリア・ピアンタドシ、フィル・ハウレット、ジョン・ボーランド。 (2007) 。 「最大の無秩序を伴うコピュラを使用したグレード相関係数の照合」、Journal of Industrial and Management Optimization、3 (2) 、305-312。
[8] Stephen Woloszynek、Zhengqiao Zhao、Jian Chen、Gail L. Rosen。 (2019) 。 「16s rRNA配列の埋め込み: 下流分析に便利なヌクレオチド配列の意味のある数値的特徴表現」、PLoS Computational Biology、15(2) 、e1006721。
[9] James K. Senter、Taylor M. Royalty、Andrew D. Steen、AmirSadovnik。 (2019) 「ディープラーニングを使用した非整列配列類似性検索」、arXiv e-prints。
[10] Ng Patrick。 (2017) dna2vec: 可変長k − merの一貫したベクター表現。 arXivプレプリント、arXiv:1701.06279。